Un Orphan Drug (OD) è un farmaco potenzialmente curativo per malattie definite rare. In realtà sarebbe più corretto il termine farmaco per malattia orfana, sia di terapia sia di una ditta farmaceutica disposta a investire molto denaro per pochi pazienti. Milioni di pazienti soffrono di malattie cosiddette rare, solo in Europa sarebbero 30milioni per più di 6.000 diverse affezioni, molte delle quali senza una cura specifica. Sviluppare OD è dunque un problema cruciale. D’altro canto, è importante anche sapere se funzionano realmente, dal momento che i requisiti per la loro approvazione sono stati finora molto deboli, a fronte di costi decisamente elevati. Purtroppo, molti medici li prescrivono solo perché sono delle novità, senza conoscerne il reale valore terapeutico. [1]
Nel 1983, il Congresso USA promulgava l’Orphan Drug Act per sollecitare la ricerca di farmaci per malattie rare, cioè con indicazioni per meno di 200mila persone. Il legislatore ha previsto allo scopo sostanziali incentivi economici, benefici fiscali e molto altro. Prima del 1983, FDA aveva approvato solo 10 farmaci con questa caratteristica, ma dal 2002 la ricerca arrivava a 1.100 molecole, di cui 232 approvate. Soltanto nel 2020 FDA ha licenziato ben 437 OD. [2] Nel 1999, anche EMA ha creato un programma simile, concedendo tra l’altro un’esclusività per 10 anni. [3] Purtroppo, sembra che l’iniziativa abbia preso una piega sbagliata. Si assiste a ciò che è avvenuto negli USA, dove l’approvazione è troppo facilitata, con prove deboli, sostenute da RCT con numeri ridotti, placebo inerte o, peggio, assenza di comparatore. Per non parlare dei costi mediamente superiori a 30mila dollari per un mese di terapia, che hanno visto molti pazienti ricorrere a prestiti e indebitarsi fino alla bancarotta. [4]
Una ricerca nata da una collaborazione fra l’Istituto Qualità ed Efficacia in Sanità di Colonia (Germania) e il Mario Negri di Milano si è recentemente occupata di OD in Unione Europea. [5] Nei primi 10 anni creazione del programma dell’EMA, fino al 2010, sono entrate in commercio 63 nuove molecole, 133 nella decade successiva; nel solo 2022 hanno visto la luce ben 22 prodotti, con un trend in continua ascesa. L’industria farmaceutica ha salutato lo sviluppo di questi prodotti come un vero successo, e così hanno fatto anche le associazioni di pazienti. La Commissione Europea ha quantificato tale successo valutando un incremento da 210 a 440mila QALY (anni di vita esenti da disabilità) per i pazienti europei.
Ma gli OD stanno rappresentando un reale progresso terapeutico? Purtroppo, non esiste una risposta convincente a questa domanda. Classificare una molecola come OD non ne accresce il valore terapeutico se mancano le prove, cioè studi comparativi con le terapie convenzionali. Solo allora l’efficacia clinica e, secondariamente, la congruità del prezzo potrebbero essere validate. EMA assegna allo status di OD numerosi benefici (Figura 1), fatti salvi alcuni criteri quali la rarità (meno di 5 casi su 10.000) e la gravità dell’affezione cui si rivolgono, e un “significativo beneficio” rispetto alle terapie esistenti (Box 1). Ma le regole adottate per l’attribuzione del “significativo beneficio” sono poco chiare. Ad esempio, per un farmaco contro l’ipertensione polmonare primitiva, la superiorità è stata assegnata soltanto con uno studio di comparazione verso placebo. In alcuni paesi i criteri di valutazione (Health Assessment Tool, HAT) sono gli stessi degli altri farmaci non orfani, dove si esaminano i reali vantaggi terapeutici. Solo in alcuni casi, però, si valuta il rapporto costi/benefici, per stabilirne la rimborsabilità. La richiesta di minori prove e la celerità di approvazione e uso da parte dei pazienti sono variabili da paese a paese. In Germania, ad esempio, lo status di significativo beneficio terapeutico si applica senza procedura HAT, con il risultato che il produttore può alzare ingiustificatamente i prezzi. Ma a che serve questa superiorità se è solo un’etichetta? La sua attribuzione, inoltre, genera ingiustificate aspettative da parte di medici e pazienti, quando in alcuni casi la “superiorità” riguarda soltanto il prezzo.
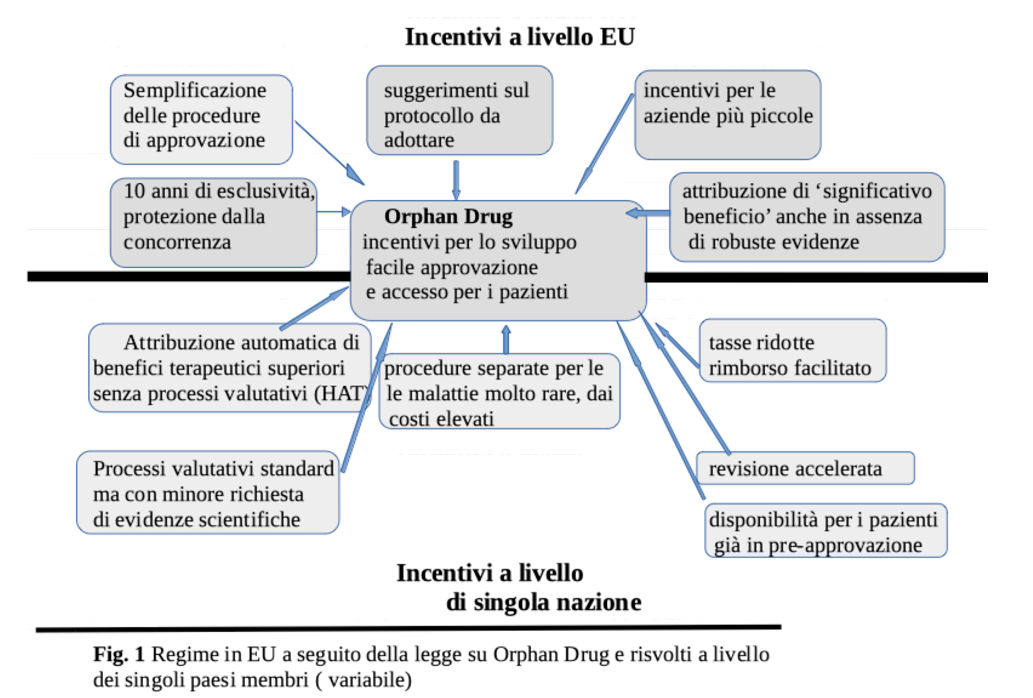

Questo è soprattutto evidente in Germania, dove i prezzi sono più alti che nel resto dell’UE, e tutti gli OD, anche quelli con beneficio incerto, sono rimborsati causando problemi di budget, con ricaduta sui prezzi in altri stati membri. Beneficio incerto e prove limitate, sono queste le caratteristiche di un OD? Dal gennaio 2011 al settembre 2021 sono entrate in commercio in Germania 89 OD, automaticamente definiti di “beneficio terapeutico superiore” (soltanto 20 dopo regolare HAT), per 78 indicazioni terapeutiche. Per queste ultime, in più della metà dei casi (54%) manca la prova di superiorità in quanto gli RCT disponibili non prevedevano un confronto con le terapie già esistenti. La carenza di meccanismi regolatori adeguati ha fatto sì che su 159 RCT relativi a 125 OD, nel 3/4 dei casi si sono usati end point surrogati (119), in 1/3 (53) dei casi mancava il gruppo di controllo e in 1/3 (50) non vi era stata alcuna randomizzione.
Gli autori dell’articolo a questo punto formulano alcune proposte operative. L’attribuzione di “superiorità” deve avvenire soltanto in presenza di prove robuste e la definizione di OD va attribuita in base all’efficacia. La superiorità va valutata nei confronti della terapia esistente, preferibilmente con RCT, anche se di esecuzione non facile, trattandosi di affezioni rare. Il problema non è insuperabile, magari ricorrendo a registri internazionali che aumentino i casi disponibili, ovvero a metodi statistici più sofisticati. Le prove più deboli andrebbero accettate solo in situazioni minacciose per la sopravvivenza, riferendosi a gruppi di controllo storici. Il confronto con lo standard di cura per gli OD è previsto solo dal 2028 in poi e la valutazione inizierà al momento dell’approvazione, anche se sono previste eccezioni, se i dati tardano a essere disponibili. Gli esperti del settore suggeriscono di attribuire la “superiorità” solo congiuntamente alla valutazione HAT, ma non è chiaro se tale suggerimento sarà seguito. Il problema principale che affligge gli OD risiede nella mancanza di solide prove che dovrebbero essere alla base dell’attribuzione di “superiorità” terapeutica e informare le decisioni circa prezzo e rimborsabilità, per la sostenibilità del sistema sanitario. L’essere OD non significa di per sé superiorità terapeutica, che dovrebbe derivare solo dal confronto con le terapie esistenti, da stabilire prima dell’approvazione ed ingresso nel mercato.
A cura di Giovanni Peronato
1. Coombs R. Rescuing orphan drugs. BMJ 2023;381:p1050
2. Ross JS. US Orphan Drug Act. BMJ 2023;381:p928
3. https://www.ema.europa.eu/en/human-regulatory/overview/orphan-designation-overview
4. Michaeli T et al. FDA approval, clinical trial evidence, efficacy, epidemiology, and price for non-orphan and ultra-rare, rare, and common orphan cancer drug indications: cross sectional analysis. BMJ 2023;381:e073242
5. Kranz P, Banzi R et al. Reforming EU and national orphan drug regulations to improve outcomes for patients with rare diseases. BMJ 2023;381:e072796